




Los priones, agentes infecciosos formados por proteínas carentes de ácidos nucleicos, causan encefalopatías espongiformes transmisibles, enfermedades degenerativas del sistema nervioso de animales y humanos.
Las encefalopatías espongiformes transmisibles, enfermedades degenerativas del sistema nervioso central, afectan a animales y humanos y son causadas por partículas llamadas priones, diferentes de otros agentes infecciosos como bacterias y virus porque carecen de ácidos nucleicos y están constituidas por una proteína de las membranas de las células.
El nombre encefalopotía espongiforme transmisible (EET) se aplica a un conjunto de enfermedades del sistema nervioso por las que el cerebro adquiere un aspecto parecido a una esponja (de allí su denominacion). Las que padecen los humanos son el kuru, la enférmedod de CreutzfeldtJokob (ECJ) el síndrome de Gerstman SträusslerScheinker (GSS) y el insomnio fatal familiar (IFF); las de los animales incluyen el scrapie de ovejas y cabras, las encefalopatías transmisibles de visones, bovinos, felinos y antilopes y la enfermedad del agotamiento crónico de mulas y ciervos en cautiverio. El scrapie del inglés to scrape, raspar por la tendencia de los animales infectados a frotarse contra postes, troncos o cercas para combatir la picazón fue reconocido en el Reino Unido, en manadas de ovejas, hace más de doscientos años. En cambio, la encefalitis espongiforme bovino (EEB), o locura bovina, apareció allí recientemente y se convirtió en epidémica; dio lugar a un serio problema de salud pública, ante la posibilidad (aún no demostrada) de su transmisión a humanos por la alimentación o por productos farmacéuticos confeccionados con material de animales infectados.
*El doctor Cumar murió el 16 de marzo de 1994
Las EET se caracterizan por su prolongado período de incubación y por su evolución, inevitablemente fatal. El examen microscópico de las zonas afectadas del sistema nervioso central revela pérdida de neuronas, cambios en otras células del tejido nervioso y aparición de unas estructuras anormales denominadas placas amiloideas, observadas en otras enfermedades, por primera vez en 1853, por el patólogo alemán Rudolf Virchow, quien les dio el poco afortunado nombre de amiloideas porque supuso que estaban compuestas por una substancia similar al almidón; posteriormente se demostró que están constituidas, sobre todo, por proteínas poco solubles en agua, cuya composición depende de la enfermedad. En los últimos años se dio un fuerte impulso al estudio de las EET al demostrarse su naturaleza infecciosa, comprobarse su transmisión accidental entre seres humanos y constatarse el contagio de bovinos por ovinos, llamado salto entre especies. En 1954, Sigurdsson llamó enfermedades provocados por virus lentos a un grupo de dolencias de origen viral caracterizadas por un período de incubación prolongado (de meses a años); ejemplos de ellas son ciertas encefalitis y el síndrome de inmunodeficiencia adquirida, sida. Durante algún tiempo se consideró que las EET eran provocadas por virus lentos no convencionales: lo primero, por su largo período de incubación, y lo segundo, por los reiterados fracasos de los intentos de aislar partículas virales en los tejidos de animales o humanos enfermos. La persistencia de estos fracasos determinó que algunos estudiosos abandonaran la idea del origen viral de las EET. Como consecuencia, hace trece años se postuló que serían producidas por agentes carentes de ácidos nucleicos, formados por proteínas, que recibieron el nombre de priones, por proteinaceous infectious particles. La reacción inicial de los científicos fue de escepticismo, pues resultaba difícil aceptar la existencia de agentes infecciosos tan diferentes de los que provocan el resto de las enfermedades transmisibles. Sin embargo, la idea de la naturaleza proteica de los priores ha ido ganando adeptos con el tiempo. Debido, sobre todo, a los estudios de Stanley B. Prusiner, de la universidad de California, en San Francisco, hoy la mayor parte de los investigadores considera que el componente infeccioso principal de un prion es una forma anómala de la proteína llamada proteino priónico, PrP (de prion protein), componente normal de las membranas celulares. La forma anormal de la PrP se designa PrPSc, para diferenciarla de la normal llamada PrPC. Recordemos que una proteína está formada por el encadenamiento de moléculas más pequeñas los aminoácidos, que se unen unas con otras mediante la llamada unión peptídico; resulta así una cadena no ramificada de aminoácidos, ordenados en una secuencia fija determinada genéticamente, que se llama estructura primaria de la proteína (véase “Proteínas a pedido”, CIENCIA HOY, 29:3142). El gen correspondiente (o que codifica) a la PrP ha sido identifcado y caracterizado, por lo que se han podido utilizar sondas de ácidos nucleicos (véase CIENCIA HOY, 20:4651) para demostrar la ausencia de tales ácidos derivados de dicho gen en los materiales infectados; ello constituye una evidencia adicional contra la naturaleza viral de los priores, ya que los virus siempre contienen ácidos nucleicos. La prueba inicial del papel desempeñado por la PrPSc se obtuvo de experimentos que demostraron que, en los cerebros de animales y humanos enfermos, existe un componente con gran capacidad de infección constituido, principalmente, por una proteína llamada PrP 2730, que está ausente en individuos sanos. Su origen es la pérdida por proteólisis (es decir por ruptura de la unión peptídica) de 67 aminoácidos de la PrPSc, durante la manipulaciones que se realizaron para purificarla. La PrP2730 es resistente a la degradación por las proteasas, que son enzimas que catalizan la ruptura de la unión peptídica. La PrPC, por el contrario, es rápidamente degradada por esas enzimas. Todas las evidencias experimentales indican que la PrPSc deriva de la PrPC. No se han advertido diferencias en la estructura primaria de ambas formas, tanto medida directamente como a partir del gen que la codifica. Sir embargo, antes de aceptar que las diferencias entre las formas normales y anormales de PrP no se deben a la alteración de dicha secuencia, se debe descartar la posibilidad de que una fracción tan pequeña que no pueda medirse de PrPSc tenga alterada su estructura primaria. Si menos del 1% de las moléculas de PrPSc se caracterizaran por aquella alteración, no serian detectables por ninguno de los métodos analíticos conocidos hasta el presente.
La descripción de las diferencias entre la PrPC y la PrPSc es uno de los campos más importantes de la investigación de los priones. Entre los posibles candidatos a explicarlas están modificaciones químicas posteriores a la síntesis de la PrP, cambios permanentes en la forma de la proteína o la unión de esta con otros componentes de las células. Las preparaciones purificadas de priones generalmente contienen otras substancias, que resultan purificadas paralelamente a la PrPSc, de las que aún se desconoce si desempeñar algún papel en la estructura y función de los priones. También las mencionadas placas amiloideas contienen otras substancias, además de la PrPSc. Se ha señalado que la PrPSc resiste a las proteasas en condiciones en que estas degradan completamente la PrPC a sus aminoácidos constituyentes. No se conoce la causa de ello, aunque tal vez consista en que la primera de ellas se una a otra molécula. Por ejemplo, la unión de proteínas con ciertos compuestos denominados proteoglicanos incrementa la resistencia de aquellas al ataque de las proteasas. Se han encontrado dos proteínas que se unen con avidez a la PrP una, sin nombre, y la otra, llamada proteína fibrilar ácido glial, un componente normal de la astroglia, variedad especial de células de sostén típicas del sistema nervioso , pero se ignora el significado de esa unión. Otra posibilidad es que tengan lugar modificaciones químicas o cambios de forma de la PrPque limiten el acceso de la proteasa a los sitios donde actúa. El cometido biológico de la PrPC es desconocido. Se ha especulado que podría desempe ñar algún papel en la regulación del número y distribución de las moléculas que reconocen la acetilcolina, una de las sustancias que transmiten información entre neuronas. Esta hipótesis, sin embargo, no es compatible con la observación de que ratones a los que se ha impedido la síntesis de PrPC conservan un aspecto y comportamiento normales, imposibles con un mal funcionamiento de los receptores de la acetilcolina. El mecanismo por el cual la PrPC se convertiría en PrPSc es también desconocido. La visión más aceptada, propuesta por S. B. Prusiner en 1982, llamada teoría del dúo mortal o de la proteína sola, parte del supuesto mencionado de que las diferencias entre PrPSc y PrPC no se deben a diferencias en la estructura primaria entre ambas proteínas, y contiene dos elementos adicionales:
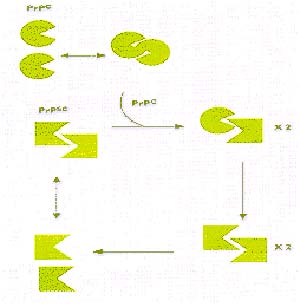
(i) que la PrPSc cataliza la conversión de la PrPC en PrPSc y (ii) que las dos formas de PrP existen como monómeros (moléculas aisladas) y dímeros (dos moléculas asociadas) en mutuo equilibrio (Fig. I)
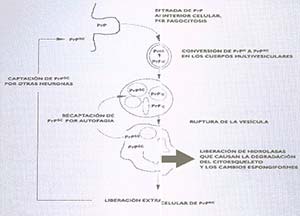
En los dímeros formados por una PrPC y una PrPSc, esta catalizaría la conversión de la PrPC en PrPSc. A medida que ello se repite, aumentaría exponencialmerte la proporción de PrPSc, de modo análogo a cómo crece, por división, un agente infeccioso convencional. La PrPSc se formaría en vesículas intracelulares llamadas lisosomas, a las que se incorpora el material fagocitado por la célula (Fig. 2).
En caso de infección, dichas vesículas pueden encerrar tanto la PrPC como la PrPSc. En un medio ácido, característico de los lisosomas, las proteínas pierden su estructura terciaria, lo cual podría facilitar la transformación de la PrPC en PrPSc. Cuando la PrPSc alcanza una concentración crítica, el lisosoma se rompe y libera la PrPSc, junto con las enzimas digestivas que contiene; ello causaría la degeneración espongiforme de las células. Producida la muerte de la neurona, la PrPSc pasaría al medio, del que sería captada por otra neurona y se reanudaría el proceso de infección y destrucción celular La teoría es compatible con resultados de estudios realizados con el microscopio electrónico, que revelan acumulación de PrPSc en la vecindad de las áreas donde se observan las lesiones espongiformes, en vesículas y otros cuerpos que contienen hidrolasas (enzimas que catalizan la hidrólisis, esto es, la escisión de sustancias en sus componentes por incorporación de agua).
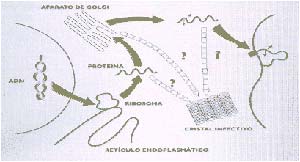
Además de la teoría de Prusiner existen otras que intentan explicar la propagación de los priones (incluyendo la viral desechada más atrás). Una hipótesis alternativa para comprender la propagación de la PrP se basa en que su forma anormal constituye estructuras cristalinas. Después de sintetizada la PrP se glicosila (esto es, se asocia con azúcares) y, posteriormente, se une con la membrana celular mediante la substancia llamada GPI (glicosidil fosfatidil inositol). La PrP es muy poco soluble en agua y, en cualquiera de las etapas posteriores a su síntesis, la forma anormal podría formar un cristal PrP infectivo, por el ordenamiento de las moléculas de PrPSc de la forma que muestra la Fig. 3.
Uno de los sustentos de esta hipótesis es que, en cerebros de animales y humanos enfermos, se observan depósitos de placas amiloideas constituidas por PrP en estado cristalino. En tal estado, podría ser resistente a la degradación enzimática, lo que explicaría esta propiedad de la PrPSc. Una vez formado, un pequeño cristal podría actuar como semilla, a la que se incorporarían moléculas idénticas que aumentarían su tamaño, en un proceso muy común en el crecimiento de cristales. Este cristal, además, podría romperse, y sus fragmentos también crecerían por acoplamiento de moléculas idénticas.
La teoría cristalina explica la multiplicación de la PrPSc en ausencia de ácidos nucleicos y define una manera de dispersión de la infección, por el traslado de fragmentos de cristales rotos. Una de las áreas más fascinantes del estudio de las EET es la de las llamadas cepas de priones, que producen enfermedades cuyas diferencias residen en su período de incubación o en la distribución del daño histológico. La existencia de esas cepas es difícil de explicar si se sostiene que los priones carecen de ácidos nucleicos; de ahí que la teoría de Prusiner intente justificar las mencionadas diferencias sobre la base de que la PrPSc puede existir en formas diversas, y que cada una de ellas actúa como molde para un determinado cambio conformacional específico de la PrPC. Por su parte, la teoría cristalina postula que esas aparentes cepas se deber a la formación de distintos tipos de cristales de PrPSc, puesto que la PrP es extremadamente heterogénea en su composición de carbohidratos; por ello, se formarían distintas clases de cristales. Un cristal está constituido por moléculas idénticas y, por lo tanto, por aquellas PrP que tienen unidos los mismos azúcares. En un intento de superar tanto las teorías virales como las no virales, y de explicar la existencia de diferentes cepas de priones, C. Weissmann propuso la llamada teoría unificada, según la cual el agente patógeno sería un holoprion, compuesto por un apoprion (formado por PrPSc), el responsable de la enfermedad y de su transmisibilidad, y por un coprion, que define las propiedades de cada cepa (Fig. 4).
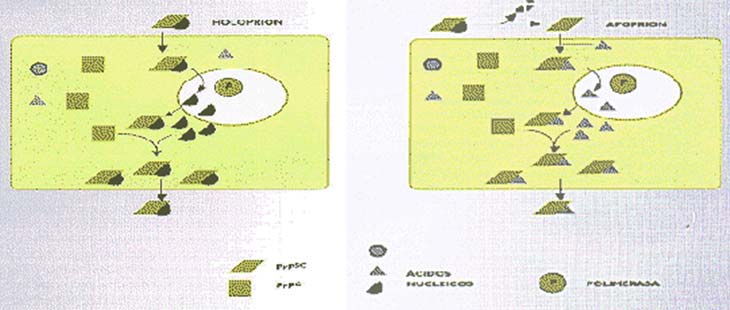
Este último estaría constituido por ácido nucleico, que podría acompañar a la PrPSc (ello nunca fue demostrado) o, por el contrario, ser un componente normal de las células no infectadas, reclutado por el apoprion y posteriormente replicado por enzimas celulares mediante un proceso estimulado por la presencia de PrPSc. Si el ácido nucleico del coprion fuera ARN, su multiplicación podría seguir un proceso parecido al del virus de la hepatitis; en tal caso, la PrPSc sería indispensable para la replicación del ARN por enzimas celulares, como sucede con la replicación del ARN del virus de la hepatitis , que requiere de la presencia del antígeno 8. Dado que la enzima responsable de ello (la ARN polimerasa dependiente de ADN) sólo puede multiplicar a ciertos tipos de ARN, se seleccionarían y amplificarían unicamente determinados ARN del coprion (véase “ADN, una molécula maravillosa”, CIENCIA HOY, 8:2635). Así se comprendería que pareciesen existir distintas cepas del agente patógeno, pues el pasaje de holopriones por diferentes huéspedes ocasionaría el cambio de los copriones.
Debido a mejoras de las condiciones sanitarias y a los avances de la medicina, la esperanza de vida en los países desarrollados pasó de unos cincuenta años, a principios de siglo, a ser hoy 73 anos para el hombre y 78 para la mujer. Pero el aumento no estuvo acompañado de una paralela mejora de la calidad de vida, pues poco se ha avanzado en el conocimiento de las enfermedades de la vejez, entre las adquieren especial relevancia las distintas formas de demencia. La definición médica de demencia, diferente de la vulgar que se refiere a cualquier tipo de enajenación mental ,incluye las alteraciones psíquicas cuya característica central es un deterioro de las funciones intelectuales superiores, una evolución crónica y progresiva y cambios de conducta y personalidad. El 4% de los estadounidenses mayores de 65 años padece alguna forma de demencia manifiesta,. el 10% presenta rasgos premonitorios de ellas, y sólo el 20% de los casos son tratables. Estas enfermedades tienen un impresionante impacto económico y constituyen un drama para las victimas y sus familias. En 1990, el gobierno de los Estados Unidos aprobó un ambicioso proyecto, denominado la década del cerebro, para entre otras cosas concientizar a la población de la magnitud del problema, promover investigaciones que favorezcan el diagnóstico, tratamiento y prevención de las demencias, incrementar los esfuerzos para lograr la rehabilitación y reinserción social de los pacientes, incorporar avances a la práctica clínica y coordinar los múltiples frentes de lucha contra estas dolencias. El 55,6% de los casos de demencia son causados por la enfermedad de Alzheimer, el 1 4,5% por infartos cerebrales múltiples, el 7,7% por el mal de Parkinson y la enfermedad (o corea) de Huntington, el 4,4% por lesiones cerebrales, el 1 2,2% por causas múItiples (depresiones, deficiencia de vitamina B12, alcoholismo crónico, trastornos metabólicos, infecciones, tumores cerebrales y otros procesos que comprimen el cerebro) y el 5,6% restante por diversas causas, entre ellas, las encefalopatías espongiformes transmisibles, que siempre dan lugar a demencias progresivas. Dado que el kuru prácticamente ha desaparecido, y que la enfermedad de CreutzfeldtJakob y el síndrome de GerstmanSträusslerScheinker son muy raros, ¿por qué dedicar tantos esfuerzos a estudiar patologías tan poco frecuentes? Muchos estudiosos piensan que su incidencia en humanos podría ser muy superior a lo que se cree, porque, frecuentemente, las EET se confunden con las enfermedades de Alzheimer de Pick o de Huntington, entre otras. Cuando se examinan materiales histológicos de pacientes del mal de Alzheimer o de demencia senil atípica, se comprueba que muchos deben ser reclasificados como enfermos de ECJ, lo que indica la necesidad de diagnósticos específicos de las enfermedades neurodegenerativas. Las EET humanas pueden aparecer esporádicamente, como lo hace el 85% de los casos de ECJ, o ser hereditarias, como el 1 0% de los de esta enfermedad y todos los de GSS y de insomnio fatal familiar. El gen que codifica la síntesis de la PrPen humanos fue clonado en 1985, luego de lo cual se comprobó en primer lugar por K. Hsiao el vinculo entre sus cambios o mutaciones y la posterior aparición de una EET; desde entonces, se han descripto catorce de dichas mutaciones, ocho vinculadas con este tipo de enfermedades. El 5% de los casos de ECJ y todos los de kuru se producen por transmisión entre seres humanos. Acerca de la primera, se ha informado sobre su transmisión por el contacto de personas sanas con material del sistema nervioso central de enfermos: por ejemplo, luego del uso de electrodos contaminados para realizar electrocorticogramas, como consecuencia de transplantes de córnea, implantes de duramadre y operaciones qui rúrgicas para remoción de craneofaringeomas. La enfermedad también se presentó en profesionales de la salud, en pacientes que recibieron hormona humana de crecimiento (hCH) y en otros tratados con gonadotrofinas hipofisiarias, hormonas extraídas de la hipófisis de cadáveres humanos. La hipófisis está situada en la base del cráneo y en estrecho contacto con el cerebro. Los casos de transmisión accidental de ECJ de mayor resonancia mundial fueron los de niños o adolescentes que recibían hCH, como tratamiento de una insuficiencia del funcionamiento de la hipófisis, trastorno que ocasiona, entre otras cosas, enanismo. En 1963 se inició en los EE.UU. y en el Reino Unido un programa de aislamiento y purificación de hGH, con el objeto tratar de manera sistemática a menores afectados por tal insuficiencia e incrementar su estatura; en 1985 se informó de la muerte por ECJ de tres pacientes del programa, lo que llevó a que, en ambos países, se prohibiera la venta de hCH obtenida de cadáveres. Mucha notoriedad publica alcanzaron casos producidos en Francia, debido a que familiares de adolescentes muertos apelaron a la justicia y obtuvieron que fueran procesados por homicidio involuntario Jean C. Job, endocrinólogo y presidente de la asociación France Hypophyse, y Fernand Dray, responsable de la producción de la hormona en el Instituto Pasteur. A partir de 1988, la hCH extraída de cadáveres humanos fue reemplazada totalmente, en los países occidentales, por una hormona sintetizada mediante procedimientos de ingeniería genética.
LAS ENCEFALITIS ESPONGIFORMES EN EL SER HUMANO
El término enfermedad de CreutzfeldtJakob fue utilizado por primera vez por Spielmeyer en 1922, luego de estudiar una mujer de veintidós años, con una rara enfermedad advertida por Creutzfeldt en 1920, y tres casos posteriores descriptos en 1921 por Jakob. Su transmisión a animales de experimentación (en este caso chimpancés) fue realizada en 1968 por Gibbs. La incidencia del mal es muy baja un caso cada millón de personas y comienza de manera lenta y progresiva, con alteraciones en la personalidad y deterioro de las funciones intelectuales y de la memoria reciente, a lo que se agregan, generalmente, contracciones bruscas y localizadas de algunos grupos musculares, las contracciones mioclónicas, frecuentemente desencadenadas por estímulos sensoriales. A medida que el mal progresa, la demencia avanza y aparecen síntomas neurológicos que indican la afectación de distintas áreas cerebrales; posteriormente se instalan signos de descerebración, que se ponen de manifiesto por la postura que adopta el paciente, hasta que entra en un estado vegetativo. La muerte sobreviene unos nueve meses después del comienzo de la sintomatología clínica. En el 60% de los afectados el electroencefalograma revela la existencia de alteraciones específicas, y si bien su diagnóstico puede realizarse con relativa seguridad a partir de los datos clínicos y electroencefalográficos, no puede considerase definitivo hasta no haber descartado otras posibles causas de demencia. La confirmación definitiva requiere una biopsia de cerebro o se obtiene en la autopsia. En 1928, Gerstmann describió por primera vez la enfermedad que luego se conoció como sindrome de GerstmannSträusslerScheinker. En 1936, dichos autores describieron siete nuevos casos, en miembros de la misma familia. El mal tiene una incidencia de uno en cada más de diez millones y la demencia, a diferencia de lo que ocurre en la ECJ, sólo aparece en sus últimos estadios, luego de una duración habitual de unos cinco años. El estudio microscópico del cerebro en la autopsia revela la presencia de placas amiloides. La transmisión a animales de laboratorio fue realizada en 1981 por Master. El kuru fue descripto por primera vez en 1956 por Gajdusek, que en 1976 recibió el premio Nobel por sus estudios de la enfermedad. La afección fue frecuente entre miembros de algunas tribus de Nueva Guinea, en cuyo idioma kuru significa temblor o estremecimiento. Publicaciones que informaron por primera vez la existencia de la enfermedad indicaban que era responsable del 50% de las muertes en algunas aldeas, y que el riesgo de padecerla era de siete a ocho veces mayor en la mujer que en el hombre. Se propagaba debido a prácticas canibalísticas funerarias, cuyo ritual tradicional establecía que mujeres y niños de la tribu (rara vez hombres adultos) comían los cerebros y otros órganos de los miembros fallecidos. La infección se producía a través de la piel o las mucosas durante la extracción y preparación de los órganos. Como no se transmite de madre a hijo, con la abolición del canibalismo, alrededor de 1956, prácticamente desapareció. Consistía en una degeneración progresiva del cerebelo. Los primeros síntomas solían ser trastornos en la coordinación de los movimientos, tan leves que únicamente el afectado los percibía, y sólo en los estadios finales llegaba la demencia. El paciente primero perdía la capacidad de caminar y, con el tiempo, no podía mantenerse de pie, sentarse, masticar ni tragar. La muerte, en la mayoría de los casos, sobrevenía a los seis meses de aparecida la sintomatología clínica. El kuru fue transmitido por Gajdusek a animales de experimentación (chimpancés) en 1966. El insomnio fatal familiar fue descripto por primera vez en 1986. Consiste en pérdida progresiva del sueño, trastornos del sistema nervioso autónomo, sudoración, fiebre, taquicardia, hipertensión arterial y alteraciones de la motricidad. A la falta de coordinación de los movimientos se agregan alteraciones del habla, contracciones musculares involuntarias y súbitas (mioclonía) y signos de daño del sistema piramidal (responsable del control, por parte del sistema nervioso central, de los músculos voluntarios). También son características de la enfermedad las alucinaciones y el estupor. La muerte sobreviene antes del año de iniciados los síntomas clínicos. El estudio histopatológico revela alteraciones severas de la corteza del cerebro, del cerebelo y del bulbo raquídeo.
Sin embargo, varios integrantes de la ex Unión Soviética aún comercializan la primera, que se consigue también en Occidente, en el mercado negro, a mitad de precio de la sintética. Sus principales consumidores son los atletas: fisicoculturistas, levantadores de pesas y corredores de velocidad y fondo, que la ingieren a pesar del riesgo, junto con esteroides androgénicos y anabólicos, para aumentar la musculatura y disminuir la masa grasa de sus cuerpos.
Los materiales bajo sospecha de estar contaminados con priones deben manejarse con extremada precaución. El departamento de Salud y Seguridad Social del Reino Unido obliga a que el descartable sea recogido en bolsas de bioseguridad de polietileno y esterilizado en una autoclave a 1341380C durante veinte a sesenta minutos, y que luego sea incinerado. El comité de cuidado de la salud de la Asociación Neurológica Americana recomienda tratar con NaOH (soda cáustica), durante sesenta minutos, a los fluidos u otros tipos de materiales infectados.
En noviembre de 1986, el laboratorio central veterinario del ministerio de Agricultura, Pesca y Alimento de Gran Bretaña informó sobre el primer caso de encefalitis bovina espongiforme (bovine spongiform encephalitis, también llamada mad cow disease, enfermedad de la vaca loca) aparecido en ese país, aunque estudios retrospectivos sugieren que otros pudieron haber ocurrido en abril de 1985; desde entonces, más de 100.000 animales han muerto allí a causa de la enfermedad, la que también fue advertida en Irlanda, Suiza, Francia, Omán y las Malvinas. Teniendo en cuenta que el período de incubación del mal es de unos cinco anos, se deduce que la infección pudo haber comenzado entre fines de la década de los setenta y principios de la siguiente.
Siempre la EEB afectó animales adultos, de entre tres y once años, que sufrieron alteraciones de comportamiento, porte y postura, y empezaron, generalmente, con signos de aprensión, ansiedad y miedo. En algunos casos, los animales enfermos patean el piso, se lamen continuamente la nariz, reaccionan desmesuradamente al sonido y al tacto y sufren notorias las alteraciones en la marcha; caminan inclinados, ejecutando pasos altos con los miembros posteriores. Los signos también pueden incluir reducción en la producción leche, debilidad, delirio y agresión (por ejemplo, son frecuentes patadas y nerviosismo general en el galpón del ordeñe). Las anormalidades en el porte se advierten en los potreros, especialmente cuando se arrea los animales o se los hace trotar porque les cuesta girar y caen con frecuencia y, en los estadios más avanzados de la enfermedad, marchan muy reclinados. El estudio histológico del encéfalo, realizado cuando el animal muere o se hace inmanejable y debe ser sacrificado, muestra vacuolización de la materia gris en el bulbo raquídeo y PrPSc en la mayor parte del las regiones del encéfalo, detectada mediante anticuerpos anti PrP.
La epidemia de EEB fue desencadenada por alimentos proteicos preparados con restos de ovejas contaminados con scrapie, enfermedad endémica en el Reino Unido. Sus inicios coincidieron con la fecha en que empezaron a aplicarse procedimientos de crianza y alimentación que favorecen a la enfermedad, debido a que el gobierno se propuso promover el aumento de la producción de leche. Para mejorar el rendimiento, los tamberos separaron tempranamente a las crías de las madres y alimentaron a estas con productos proteicos derivados de carne, hueso y otros despojos de rumiantes. Con el fin de preparar dichos productos, se utilizaba hexano, como solvente que eliminara los lípidos es decir las grasas del material ovino. Dado que la PrPSc es poco soluble en agua y muy soluble en hexano, era extraída de los despojos en las etapas iniciales de esa deslipidización; pero una de las crisis petroleras llevó a suspender el uso del hexano, con lo que se perdió la capacidad de eliminar la PrPSc
En los momentos iniciales del consumo de alimentos contaminados, la transmisión de la enfermedad de ovinos a bovinos fue poco importante, pero luego de un tiempo se declaró la epidemia, porque se produjo un cambio en la forma de conversión de la PrPC en PrPSc. En los primeros bovinos infectados, algunas pocas veces la PrPSc del ovino actuaba como catalizador y se formaba PrPSc bovina; a medida que esta se acumulaba, la conversión de PrPC en PrPSc se hizo más eficiente y rápida, con la consiguiente aceleración de la diseminación infecciosa.
Las vacas locas que murieron a causa de la EEB generaron uno de los mayores problemas de la ganadería británica moderna, que afectó en primer lugar a la economía pero, también, amenaza a la salud pública humana, pues la naturaleza de la enfermedad y el tipo de tejido afectado hacen temer que acontezca la transmisión de bovinos a humanos, como ocurrió de ovinos a vacunos. Existe, entonces, preocupación por inactivar eficientemente a los priones que pudieran estar presentes en materiales destinados a industrias como la alimenticia y la farmacéutica la segunda, por ejemplo, en muchas ocasiones obtiene productos a partir de extractos de órganos animales. En los EE.UU. y en la Unión Europea no se permite vender fármacos de procedencia animal o humana si los procedimientos de su elaboración no satisfacen ciertos requisitos considerados efectivos para eliminar la posible contaminación con priones.
En líneas generales, dichos requisitos derivan de lo siguiente:
Puesto que la EEB se manifesta en bovinos de cinco años, y se ha determinado una fase preclinica en animales de unos diez a doce meses, es aconsejable usar ejemplares de menos de ocho meses para la obtención de los productos biológicos.
Es conveniente utilizar los tejidos o fluidos de menor riesgo. Los de máximo riesgo son el cerebro, el cordón espinal, el bazo, los nódulos linfáticos, el colon, los nervios, la pituitaria, las glándulas adrenales y liquido cefalorraquídeo; los de riesgo intermedio, el timo, el pulmón, el corazón, el músculo esquelético, los riñones y las tiroides; los de riesgo mínimo, la orina, las heces y otros fluidos corporales.
Es deseable usar animales de zonas libres de EET. Australia y Nueva Zelandia son los únicos países clasificados por la OIE (Organización Internacional de Epizootias) como libres de scrapie y EEB.
Es preferible implantar controles de calidad en la elaboración de productos de origen biológico, que permitan determinar la presencia de PrPSc en la materia prima y el producto frial.
PROCEDIMIENTO DESARROLLADO LOCALMENTE PARA EVITAR LA CONTAMINACIÓN POR PRIONES
Desde 1987, el CIQUIBIC (Universidad Nacional de CórdobaCONICET), lugar de trabajo de los autores, ha celebrado varios convenios de transferencia de tecnología con un laboratorio comercial de especialidades medicinales de Buenos Aíres. La finalidad fue definir métodos para obtener gangliósidos (un tipo particular de lípido de las membranas celulares) a los que se les asigna capacidad de estimular el desarrollo y la nutrición de las neuronas y que podrían ser útiles para contrarrestar los efectos adversos de una serie de afecciones del sistema nervioso. Dado que los gangliósidos en cuestión se extraen de cerebros bovinos, el laboratorio buscaba eliminar su posible contaminación con priones y, además, necesitaba un procedimiento de análisis que permitiera detectar la PrPSc.
Los autores desarrollaron un método novedoso, patentado en 1992 en la Argentina con el nombre Proceso para obtener gangliósidos puros libres de contaminación debida a agentes infectivos no convencionales. Su aplicación en el proceso de obtención de gangliósidos no afecta el rendimiento industrial ni la calidad farmacéutica de los productos. El mismo año, el Instituto di ricerche biomediche Antoine Marxer, de Italia (uno de los tres reconocidos para realizar tales estudios por la Food and Drug Administration y los National Institutes of Health de los Estados Unidos), sometió el procedimiento a verificación experimental en animales de laboratorio, único método de reconocida eficacia para demostrar la ausencia de los contaminantes que se buscó eliminar. Los resultados del experimento, enviados al European Journal of Veterinary Pathology, demuestran que es posible obtener gangliósidos que mantienen sus propiedades químicas y biológicas y que están libres de infectividad, aun cuando el material de partida esté contaminado.
Es necesario asegurarse que los procesos de extracción y purificación sean capaces de remover o inactivar los priones, lo que se verifica mediante ensayos del producto purificado.
Son cada vez mayores las exigencias que gradualmente se están imponiendo para garantizar la ausencia de priones en animales aptos para el consumo humano. En la industria farmacéutica, ya sólo son aceptables órganos de animales enteramente sanos. De allí la importancia económica de demostrar que los animales o sus productos que se comercializan están libres de infección por priones.
Pablo E. A. Rodríguez
Universidad Nacional de Cordoba CONICET
Federico A. Cumar
Universidad Nacional de Cordoba CONICET




