




En la enfermedad de Alzheimer, que afecta gran parte de la población de más de 65 años, se observa degeneración del rejido nervioso cerebral. Las manifestaciones clínicas características son la pérdida progresiva de la memoria, dificultad en el aprendizaje, desorientación, cambios de personalidad, etc. En este artículo se explican los cambios bioquímicos que ocurren en el cerebro de los enfermos de Alzheimer, probablemente, responsable, de las alteraciones mentales que estos evidencian.
Entre las investigaciones que tratan de entender el origen y la evolución de la enfermedad de Alzheimer se destacan aquellas que abordan los mecanismos bioquímicos implicados en esta dolencia.
La enfermedad de Alzheimer es la causa más frecuente de demencia entre las personas mayores de 65 años y es, probablemente, la patología cerebral más estudiada desde el punto de vista bioquímico y genético en la última década. Los estudios estadísticos muestran que una de cada veinte personas entre los 50 y 70 años padece esta enfermedad; entre los mayores de 85 años el número de afectados asciende al 20%. Se estima que la situación se agravará en un futuro cercano, ya que como la expectativa de vida en los países desarrollados supera los 75 años, para el año 2000 el 20% de la población rondará los 65 años. Es decir que, en pocos años más, una alta proporción de la población adulta puede adquirir esta afección, que es de alto costo familiar y social.
Es esta una enfermedad de evolución lenta, y se caracteriza por la pérdida progresiva de la memoria, la orientación, el juicio y el lenguaje. En promedio, su duración es de 8 a 12 años, en los que existe un período de 2 a 3 años en el que la sintomatología es muy sutil y muchas veces pasa inadvertida. A pesar de disponer de protocolos de diagnóstico clínico cuidadosamente diseñados, la certeza del diagnóstico de la enfermedad de Alzheimer es de aproximadamente 85%, y sólo se confirma por el examen postmortem del cerebro.
El factor de riesgo más importante asociado con la enfermedad es la edad, ya que el cerebro presenta cambios estructurales y funcionales durante el envejecimiento. Recordemos que el tejido nervioso está compuesto por distintos tipos de células: las neuronas y la neuroglia, que incluye a los oligodendrocitos, los astrocitos y las células de la microglia. Las neuronas parecen ser las células más sensibles a los efectos del envejecimiento, ya que con el tiempo se modifica tanto su cantidad como su morfología. A partir de los 50 años de edad se pierden alrededor del 5% de neuronas por cada diez años de vida, y aparecen alteraciones similares a las que se han descripto en la enfermedad de Alzheimer en áreas del cerebro que son fundamentales para las funciones cognitivas. Sin embargo, existen diferencias cualitativas y cuantitativas entre los cerebros con envejecimiento normal y los enfermos. Alois Alzheimer (véase el recuadro biográfico “Alois Alzheimer”) fue el primero en asociar estas lesiones con la demencia, ya que en 1907 publicó un informe en el que presentaba la descripción del cerebro de una paciente afectada de demencia a edad temprana. A partir de ese momento, se definió anatómica y clínicamente la enfermedad que lleva su nombre (véase “Demencia y Depresión”).
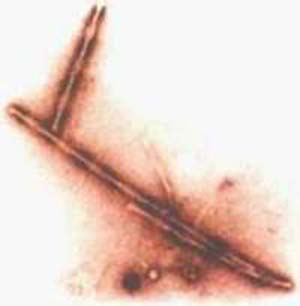
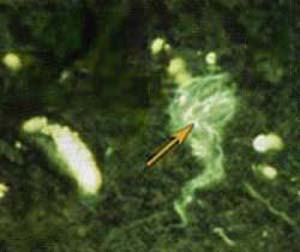
El examen microscópico de cortes de cerebro de los enfermos de Alzheimer revela pérdida neurona y la presencia de las dos alteraciones típicas de esta enfermedad: la degeneración u ovillo neuro- fibrilar y la placa neurítica, también llamada placa senil. Estas lesiones se localizan principalmente en la corteza cerebral asociativa y en el hipocampo; en los casos avanzados reemplazan gran parte del tejido cerebral normal. Aunque existen discrepancias sobre cuál de las dos lesiones es la más importante en el desarrollo de la enfermedad, existe buena correlación entre el grado de demencia y el número de ambas lesiones presentes en el cerebro. La degeneración neurofibrilar es una lesión intracelular que afecta principalmente a las grandes neuronas piramidales. Se considera que es una respuesta a distintos estímulos agresivos, o sea que se puede presentar en diferentes patologías. Esta lesión aparece como un ovillo compuesto por fibrillas entrelazadas, muy insolubles en agua, de 20 nm de espesor, conocidas como filamentos apareados helicoidales (Figuras 1 y 2). Su principal constituyente es la proteína tau, normalmente asociada a los microtúbulos que forman parte del citoesqueleto y del sistema de transporte de sustancias dentro de las neuronas. En la enfermedad de Alzheimer la proteína tau se modifica por agregado extra de grupos fosfatos, los cuales alteran su solubilidad y su unión con los microtúbulos (véase “Importancia de la Proteína Tau en la enfermedad de Alzheimer”).
Las placas neuríticas son estructuras esféricas, relativamente grandes (10-200 µm), que se ubican entre las células (Figura 3); presentan una zona central compacta rodeada de prolongaciones neuronales alteradas o dañadas, astrocitos y células de la microglia. El componente principal de la zona compacta es una sustancia insoluble conocida como amiloide beta (Ab ) que se deposita también en las paredes de las arteriolas, venas y capilares del cerebro. Aunque la presencia del amiloide en las placas neuríticas fue detectada hace 70 años, recién en la última década se ha logrado su caracterización bioquímica.
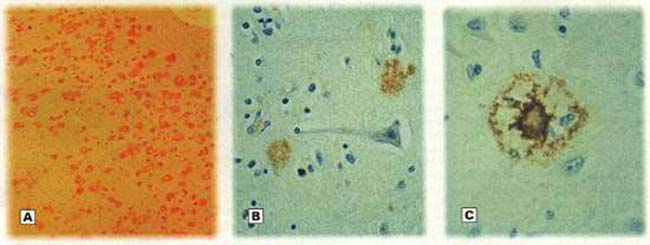
Entre 1984 y 1985 se demostró que el Ab es un péptido compuesto por 40-43 aminoácidos; aunque su secuencia -es decir, el orden en que están ubicados los aminoácidos- no se parecía a la de ninguna otra proteína conocida hasta el momento, se observó que el Ab se comportaba de la misma forma que las proteínas amiloides conocidas, es decir, que formaba fibras insolubles (véase “Las proteínas amiloides”).
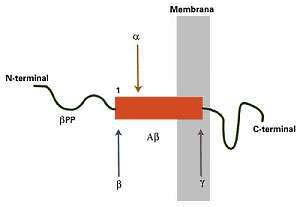
El Ab es parte de una proteína de mayor tamaño, denominada bPP (por b amyloid precursor protein), de la cual no se conoce la función biológica, aunque se sabe que forma parte de las membranas de todas las células del cerebro y de otros tejidos. La bPP puede ser digerida por lo menos por tres tipos distintos de enzimas celulares denominadas secretasas a, b y g. La acción conjunta de las b y g secretasas Iibera la secuencia de aminoácidos que corresponden al Ab , por lo cual este proceso se denomina amiloidogénico (Figura 4). Por el contrario, la a secretasa corta la proteína precursora de tal manera que no se libera el amiloide beta, originando así la ruta no amiloidogénica (Figura 4).
Es decir que tanto el péptido Ab intacto así como sus fragmentos se encuentran en el plasma y líquido cefalorraquídeo de personas normales, aunque hasta el momento se desconoce si estas moléculas poseen alguna función biológica. Sin embargo, existe una diferencia importante entre este Ab presente en los fluidos biológicos, constituido por 40 aminoácidos (lo llamaremos Ab corto), y el que forma parte de las placas neuríticas; en este caso la especie mayoritaria de Ab tiene 42 aminoácidos (Ab largo).
Uno de los problemas que enfrentan los investigadores es por qué y cómo el péptido Ab , que es un producto normal de las células, deja su estado soluble para agregarse ordenadamente en forma de fibras insolubles que se depositan masivamente en el cerebro de los enfermos de Alzheimer. Diversos factores parecen tener importancia en esta transición:
* longitud del péptido: experimentos realizados in vitro han demostrado que el péptido Ab largo es más hidrofóbico -rechaza el agua- y forma fibras insolubles más rápidamente que el corto. Se ha observado, además, que si se agregan fibras formadas por el Ab largo a cultivos de neuronas se induce la muerte de estas, o sea que la acumulación de la forma larga del Ab en el tejido cerebral resultaría más tóxica para las neuronas que la acumulación de la forma corta. Existen evidencias experimentales recientes que sugieren que el desbalance hacia una mayor producción celular de la forma larga del Ab sería un hecho importante en el desarrollo de la patología de la enfermedad de Alzheimer.
* concentración y pH: utilizando el péptido Ab sintético se demostró que tanto la cantidad como la velocidad de la formación de la fibra es directamente proporcional a la concentración de péptido, y que se ve favorecida en el rango de pH fisiológico.
* modificaciones de los aminoácidos: el análisis bioquímico del amiloide extraído de las placas neuríticas reveló que algunos de los aminoácidos que forman parte de su estructura tienen modificaciones químicas; por ejemplo, se ha detectado isomerización -es decir, cambios en la configuración- y ciclización del ácido aspártico ubicado en el extremo amino terminal. Queda aún por ver si estas alteraciones son previas al depósito de Ab o son modificaciones que ocurren una vez que este se ha depositado como fibra.
* sustituciones de aminoácidos: el reemplazo de un aminoácido por otro en la secuencia del Ab puede ser crucial en su capacidad de formar fibras. Se ha demostrado que la presencia de glutamina en reemplazo de ácido glutámico en la posición 22 incrementa enormemente la tendencia de A13 por agregarse, aunque estos dos aminoácidos se diferencian solamente en un grupo NH2, presente en la glutamina y no en el glutámico.
* interacción con metales: tanto los iones zinc (Zn) como aluminio (Al), en concentraciones fisiológicas o ligeramente superiores, inducen la agregación en forma de fibras de péptidos sintéticos de secuencia idéntica al Ab . Se sabe que el Al se acumula en el cerebro durante el proceso normal de envejecimiento. Puesto que el Al se encuentra presente en las placas neuríticas y en los filamentos apareados helicoidales, algunos investigadores postulan un rol fisiopatológico de este metal en la patología de Alzheimer. Una de las hipótesis sugiere que, además de interactuar con el Ab , el Al podría unirse a la proteína tau hiperfosforilada, contribuyendo así a su insolubilización. Sin embargo, los estudios epidemiológicos no han podido confirmar aún una relación directa entre el Al y la enfermedad que nos ocupa (véase “Los radicales libres en la enfermedad de Alzheimer”).
* interacción con otras macromoléculas: el efecto tóxico del amiloide sobre las neuronas estaría determinado por su capacidad de formar fibras y la resistencia de estas a la proteolisis (digestión por enzimas especificas, denominadas proteasas). Se han descripto proteínas asociadas a los depósitos de Ab que facilitarían alguno de estos dos procesos; por ejemplo, el componente amiloide P y los proteoglicanos protegerían al Ab de la proteolisis, mientras que la a 1-anti quimiotripsina y la acetilcolinesterasa facilitarían la formación de fibras.
La enfermedad de Alzheimer puede ser esporádica o familiar. La primera es la más frecuente, ya que corresponde al 80-90% de todos los casos detectados. Esta forma no está asociada a defectos genéticos y generalmente ocurre después de los 65 años de edad. Sin embargo, aunque la mayoría de los casos sean esporádicos, el estudio de las formas hereditarias ha sido de gran importancia para el estudio sistemático del rol de algunas de las proteínas involucradas en la enfermedad.
La enfermedad de Alzheimer familiar o hereditaria se presenta en dos formas distintas: la de inicio temprano -comienza antes de los 65 años- que es de curso rápido, evolución agresiva, y se transmite en forma dominante, y la de inicio tardío -se manifiesta después de los 65 años- que es menos agresiva, de evolución lenta y que presenta un patrón de herencia más complejo.
Los estudios genéticos realizados en miembros de diferentes familias afectadas por la enfermedad de inicio temprano indicaron que había alteraciones en los genes correspondientes a la proteína precursora del Ab, el bPP, y otras dos proteínas denominadas presenilina 1 y presenilina 2. El hecho de que las personas afectadas por el síndrome de Down -los cuales tienen una copia adicional del gen del bPP- presentan lesiones cerebrales idénticas a los enfermos de Alzheimer cuando viven más allá de los 40 años, permitió asociar las mutaciones en el bPP con el desarrollo de la enfermedad. Las presenilinas 1 y 2 son proteínas de membrana, muy semejantes entre sí, constituidas por 467 y 448 aminoácidos, respectivamente; en ambos casos la cadena de aminoácidos atravesaría la membrana de 7 a 9 veces (véase la Figura 5) y, en el cerebro, se localizan en el cuerpo de las neuronas y en las dendritas, excluyéndose los axones. Se conoce muy poco sobre la biología celular de estas proteínas, ya que han comenzado a estudiarse en los últimos dos años.
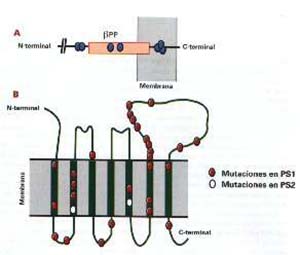
Hasta el momento se han detectado entre 5 y 7 mutaciones patogénicas en el gen del bPP y más de 30 en las presenilinas (Figura 5), las que se heredan en forma dominante, no ligada al sexo, y son causa suficiente para provocar la enfermedad de Alzheimer a edad temprana en familias de diverso origen étnico.
Así, un 60-63% de la población es homozigota para el alelo E3 -o sea, que lleva este gen en ambos cromosomas- mientras que los heterozigotas E3/E4 representan el 20-23% y los E2/E3, el 11-12%. Las otras combinaciones de genes de la Apo E -o sea, E2/E4, E2/E2 y E4/E4- se encuentran en el 1-3% de las personas.
Los resultados de numerosos estudios realizados indicaron que la frecuencia del alelo E4 en la población afectada por la forma tardía de la enfermedad Alzheimer es hasta cuatro veces mayor que en los controles (personas no afectadas), y que estos valores son independientes del origen étnico de la población estudiada. Debido a que existen portadores del alelo E4 que no desarrollan la enfermedad, y a que también hay pacientes en los que el alelo no está presente, se concluyó que el alelo E4 de la Apo E no es un gen causal de la enfermedad, pero sí un factor de riesgo genético importante, no sólo en grupos familiares con la forma de la enfermedad de inicio tardío, sino también en un elevado porcentaje de casos esporádicos. Se estima que el riesgo es de 3 a 4 veces mayor para los portadores de un alelo E4, y de 12 a 16 veces mayor para los homozigotas.
Si bien en los últimos años se avanzó en el estudio de la genética de las formas familiares de origen temprano del Alzheimer, existen muchos casos no relacionados con ninguno de los genes mencionados. Además, se han descripto casos de inicio precoz sin historia familiar, lo que sugiere que podrían estar involucrados otros genes aún no localizados o factores genéticos que actúen en forma sinérgica entre sí, o con factores ambientales, para iniciar la enfermedad. Quedan aún por identificar los genes y/o los factores ambientales causantes de la mayoría de los casos esporádicos de inicio tardío y de un significativo porcentaje de los casos de inicio precoz para explicar en su totalidad esta enfermedad. Es importante señalar que no se han encontrado asociaciones genéticas entre los pacientes familiares o esporádicos y el gen que codifica la proteína tau.
Se ha demostrado que todas las mutaciones genéticas asociadas a la enfermedad de Alzheimer estudiadas hasta la fecha tienen una repercusión bioquímica detectable, lo cual podría explicar, al menos en parte, el desarrollo de la enfermedad. Los efectos de estas mutaciones han sido mayormente relacionados con la formación de amiloide (véase la Figura 6). Por ejemplo, si se introduce en células en cultivo el gen del bPP mutado cerca del extremo carboxilo terminal correspondiente al Ab, se produce mayor cantidad de la forma larga del Ab que en las células en que se incorporó el gen normal. Estos datos se correlacionan con lo que ocurre en humanos, ya que las células de los individuos portadores de dicha mutación también producen más péptido Ab largo que las células normales. Por otro lado, el tejido cerebral de individuos afectados con esta enfermedad presenta mayor cantidad de la forma larga del péptido Ab en comparación con cerebros normales provenientes de ancianos. Este fenómeno es más evidente aún en los casos de la forma familiar de comienzo precoz. La hipótesis actual propone que las mutaciones en el bPP cercanas al carboxilo terminal de Ab alterarían la acción de la g secretasa, originando una mayor producción del Ab largo con respecto al corto.
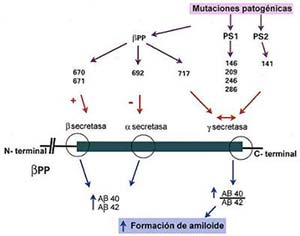
El signo (+) indica mayor producción de Ab40 (forma corta del amiloide) y Ab42 (forma larga) por edecto de bsecretasa, (-) mayor producción de Ab40 y Ab42 por menor acción de a secretasa y (<--->) modificación del sitio de corte en el extremo carboxilo terminal de ab que lleva a mayor producción relativa de Ab42.
También se ha demostrado que en los pacientes que presentan mutaciones en los genes de las presenilinas se produce mayor cantidad de la forma larga del amiloide. Se postula que, por efecto de las mutaciones, las presenilinas interferirían en el tránsito intracelular del bPP y alterarían su procesamiento enzimático, especialmente en el sitio de acción de la g secretasa.
Si bien no se conoce la relación entre el bPP y las presenilinas, se ha demostrado recientemente que ambas moléculas jugarían un papel importante en la reacción de las neuronas sometidas a situaciones de estrés, como lo son la deprivación de factores neurotróficos, el daño oxidativo o el depósito de amiloide. Las neuronas, como todas las células, pueden morir siguiendo dos procesos diferentes: la apoptosis o la necrosis. La apoptosis, también llamada muerte celular programada, implica un proceso celular activo, controlado genéticamente, que es sencillo de diferenciar de la necrosis -simplemente, el estallido celular- mediante criterios morfológicos y bioquímicos. Este proceso es el resultado de la expresión de distintos genes, algunos de los cuales funcionan como promotores y otros como inhibidores. Los estudios, realizados in vitro, sugieren que el bPP y la presenilina 2 actuarían regulando mecanismos de muerte celular por apoptosis. Las mutaciones en los genes de estas proteínas parecen alterar groseramente dicha regulación, provocando un aumento de la muerte de las neuronas. Desde el punto de vista de la patología, si la pérdida de neuronas en el Alzheimer está mediada por un mecanismo programado, resultaría posible -al menos en teoría- controlarlo genéticamente, o bien modularlo con factores de crecimiento específicos, a fin de rescatar neuronas afectadas.
Con respecto a los posibles mecanismos por los cuales el alelo E4 de la Apo E se relacionaría con la enfermedad de Alzheimer, existen diversas líneas experimentales:
* se ha demostrado que Apo E se une al Ab en los depósitos cerebrales y que esta unión podría modular la formación de fibras. Es posible que este efecto sea diferente para cada isoforma, es decir, para la proteína que se originó por cada alelo. Es importante remarcar que los cerebros de pacientes homozigotas para E4 tienen más cantidad de amiloide depositado que los que no poseen dicho alelo.
* en los cultivos de neuronas, Apo E posee un efecto promotor del crecimiento de prolongaciones nerviosas, pero Apo E4 sería menos eficiente que Apo E3 o Apo E2. Estos resultados posiblemente puedan asocíarse a una de las principales funciones bioquímicas de Apo E en el sistema nervioso, la de redistribuir colesterol para reparar membranas dañadas.
* se ha demostrado recientemente que las distintas isoformas de Apo E poseen diferentes efectos como moléculas anti-oxidantes; Apo E4 sería menos efectiva que Apo E2 y Apo E3 en proteger a las neuronas del daño oxidativo.
Por el momento, no existe un tratamiento eficaz para la enfermedad de Alzheimer, ya que es imposible detener o revertir el depósito masivo de proteínas fibrilares que producen el daño neuronal. Las estrategias experimentales con objetivos terapéuticos se centran en tres puntos clave: disminuir la secreción de la forma larga del Ab por las células del sistema nervioso central, interferir en su agregación, y controlar la reacción inflamatoria que surge en el cerebro como respuesta al depósito de Ab . Esta reacción se debe a la presencia, en el entorno de la placas neuríticas, de células de la microglia activadas, las que producen moléculas pro inflamatorias y factores que contribuirían al daño neuronal. (Véase “¿Quién es, en realidad, el culpable?”.)




